- Liittynyt
- 30.10.2016
- Viestejä
- 2 616
No jos esim. kaikilla sinisilmäisillä on yhteinen kantaisä ja siellä todennäköisesti ei ole yhtään tai kovinkaan paljoa ruskeasilmäisiä joukossa koska ruskeasilmäisyys on dominoiva ominaisuus niin kyllä se aika sisäsiittoista meininkiä on.
Ei toimi noin.
Sinisilmäiset eivät katoa
Ruskea silmien väri on vallitseva piirre. Tarkoittaako se sitä, että ennen pitkää kaikista tulee ruskeasilmäisiä?
tiistai 24. tammikuuta 2012
#silmät #Kysymykset #silmien väri
Ei tarkoita. Ihmisellä on useita eri geenejä, jotka ohjaavat pigmentin eli väriaineen muodostumista iiriksen eli silmän värikalvon stroomasoluissa.
Silmien värin periytymisen monimutkaisuus käy ilmi jo, kun tutkitaan, miten pelkästään ruskean värin tuottava geeni periytyy.
Geeneistä esiintyy vaihtoehtoisia muotoja eli alleeleja, jotka voivat olla toimivia tai toimimattomia. Ruskeaa iiriksen pigmenttiä tuottavan geenin sanotaan olevan vallitseva, koska lapsesta tulee ruskeasilmäinen, vaikka hän saisi geenin toimivan alleelin vain toiselta vanhemmaltaan.
Toimimattomia ja toimivia geenejä
Jos lapsi perii molemmilta vanhemmiltaan toimimattoman alleelin, ruskeaa pigmenttiä ei synny, vaan iiris jää siniseksi. Ruskeasilmäiselläkin voi siis olla perimässään alleeli, joka johtaa lapsen sinisilmäisyyteen.
Silmien värin jakauma ei muutu väestössä
Geenin vallitsevuus ei siis tarkoita sitä, että se syrjäyttäisi muita geenejä. Toimimattomat geenialleelit pysyvät osana perimää, elleivät ne heikennä lajin säilymistä. Iiriksen pigmentti ei ole lajin säilymisen kannalta ratkaiseva ominaisuus. Ihmisten silmien väri vaihtelee tulevaisuudessakin, ja todennäköisesti värien keskinäiset suhteetkin pysyvät suunnilleen samoina.
Alleelien yhdistelmä määrää silmien värin
Ruskeaa iiriksen pigmenttiä tuottaa geeni EYCL3 ja vihreää geeni EYCL1. Kummastakin on kaksi eri muotoa eli alleelia.
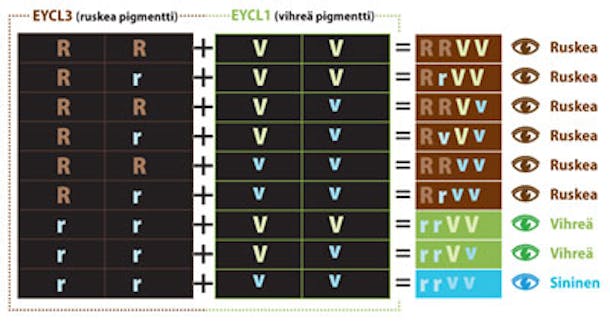
R ja V tuottavat runsaasti pigmenttiä, ja heikommat alleelit r ja v tuottavat vain vähän pigmenttiä.
R on vallitseva V:hen nähden, ja V on vallitseva r:ään ja v:hen nähden. Lapsi saa yhden alleelin kummaltakin vanhemmaltaan. Jos hän perii molemmilta heikon alleelin, pigmenttiä syntyy vain vähän ja silmät näyttävät sinisiltä.
Pigmenttikasat tuottavat harmaat silmät
Silmien väri ei riipu vain iiriksen pigmentistä vaan myös pigmentin määrästä ja jakautumisesta iiriksessä. Tämä koskee erityisesti muita värejä kuin ruskeaa, vihreää ja sinistä.
Yhden teorian mukaan harmaasilmäisillä on samat pigmentit kuin sinisilmäisillä, mutta heillä pigmentit ovat kasaantuneet isommiksi joukoiksi, jotka heijastavat valoa eri tavalla kuin tasaisesti jakautuneet värihiukkaset. Silmien väriin vaikuttavat siis myös ne geenit, jotka säätelevät pigmenttien sijaintia iiriksessä.
")






